This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
He also discussed the importance of having retention samples for raw materials and packaging materials and the role of quality control in investigating deviations and ensuring the stability of raw materials. The panel also discussed the role of analytical methodvalidation when outsourcing testing and the necessity of Excel sheet validation.
Therefore, extractables and leachables assessment should consider the packaging components of the CCS, including the labelling. However, according to the FDA, this is “less of a concern” for products, such as biological products, that are packaged in glass containers.
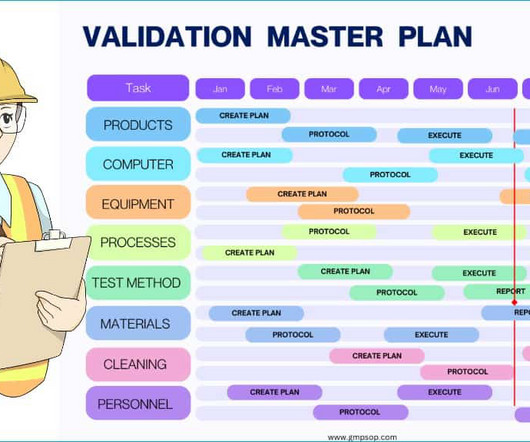
In general, you must formally validate all process steps, production equipment, cleaning methods, testing methods, computer systems and environment, facilities and utilities which are directly used for the manufacture of sterile and non-sterile products. This validation is relatively less comprehensive.
Following are some examples of validation studies which should be included in the validation master plan: 1. Process validation master plan The process validation master plan accounts for all manufacturing and packaging processes directly employed in the manufacturing and packaging registered products.
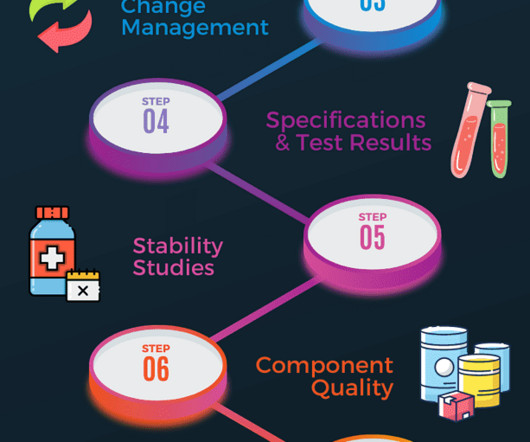
Component quality A review of all starting materials and primary packaging in contact with the product should be completed. This will include supplier performance assessment and identifying any critical deviations associated with active or excipient ingredients, primary packaging and closure material. Subscribe f.
Validated test method A test method comprehensively describes all procedures used in sample analysis. Analytical test methodvalidation guarantees that your test method is robust enough to provide evidence if your product meets the predetermined product specification or conforms to the failure of a product.
Stability testing overview for Pharmaceutical products Pharmaceuticals quality assurance & validation procedures GMPSOP %title% Prev PREVIOUS POST Pharmaceutical products must maintain quality, safety, purity and efficacy throughout their specified shelf-life conditions up to their nominated expiration date.
Laboratory documentation and records that should be available include: – Test methods and test reports – Laboratory notebooks/sheets, instrument records, and calculations – Conditions of tests and instrument settings. – Test methodvalidation protocols, data and reports – Other records and data i.
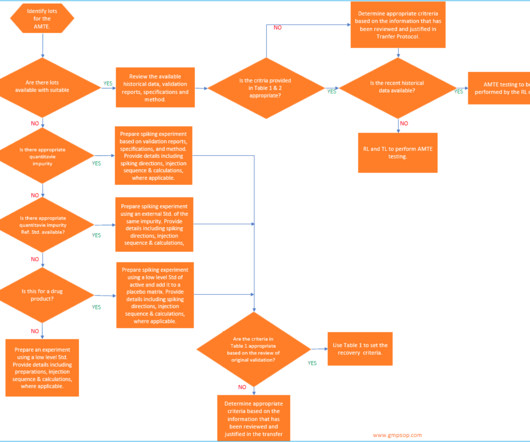
According to USP <1224> , method transfer is defined as the documented process that qualifies a laboratory (RL) to use an analytical method originating from another laboratory (TL), regardless of whether it’s internal or external. 240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms.
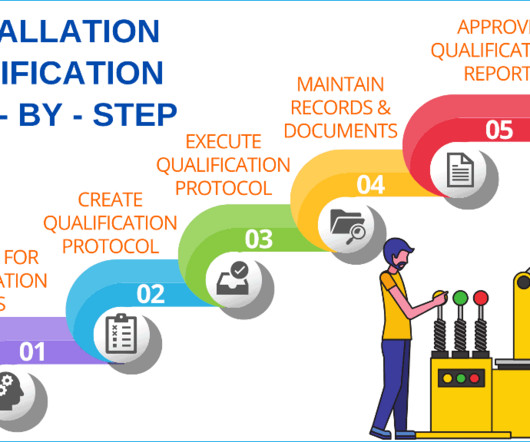
Add the objective of the installation qualification protocol Write the objective of the protocol defining the installation qualification (IQ) and operational qualification (OQ) requirements and acceptance criteria for the equipment with location i.e., packaging or manufacturing, and the facility.
If my cleaning methods are not validated would they cause contamination of my products? What is the probability my cleaning methods weren’t validated? Should I implement a more robust cleaning methodvalidation to mitigate such risk? Packaging and labelling operations v. Manufacturing processes iv.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













Let's personalize your content