This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The rules were implemented to enhance patient safety protections via revised drug handling, packaging, and delivery requirements. The rules include changes to the notification process, medication packaging, the handling of reports, and safety issues. Hyman, Phelps & McNamara, P.Cs
This document is revised from a version published in October 2023. Firstly, leachables can compromise a products quality and therapeutic effect, by potentially interacting with the formulated drug product, the document reported. Moreover, manufacturers should documentinformation about their safety thresholds, the guidance stated.
One area in which the problem can be tackled effectively is product packaging. With predictions that the global market for anticounterfeiting packaging is set to reach almost $250 billion by 2026, growth in authentication and anti-tamper devices such as holograms appear to have a healthy future.”
Clinical packaging and labelling follow stringently controlled procedures and high-standard quality control measures to assure the safety and functionality of investigational medicinal products, during their storage, distribution, and use. Finding the best clinical trial packaging services providers. Trends in clinical packaging.
Packaging plays a vital role in maintaining the quality, safety, user-friendliness and marketability of drugs and other pharmaceutical products. Finding the best commercial packaging suppliers in contract marketing. Pharmaceutical packaging formats and materials. Pharmaceutical packaging formats and materials.
Proper documentation and effective data management were underscored as essential elements in building and sustaining a culture of quality and compliance within the industry. Embracing innovation in pharma packaging is vital become future Shivaji Chakraborty, Asst.
The cobas pulse system is a point-of-care device that combines a glucose test strip reader with a touch screen that is used for displaying patient information and data, as well as analysing and sharing clinical results.
Particularly the printed packaging materials where product information is presented. Printed packaging materials typically include product names, active ingredients, concentration, batch numbers, expiry dates, registration numbers, barcodes, etc. Additional documents included each month. Checkout sample preview s.
Table of Contents Good documentation practice requires a system for control, implementation, maintenance and archival of GMP related documents and records. To implement good documentation practice and systems the site must assure that GMP documents and records are adequate, approved and in compliance with applicable GMP requirements.
As part of these efforts, the government has implemented regulations mandating API (active pharmaceutical ingredient) manufacturers and pharma brands to incorporate barcoding on product packaging. This regulatory step is aimed at preventing falsified medicines from entering the market.
To maintain the integrity and safety of these investigational products, you must adhere to Good Manufacturing Practice (GMP), FDA Clinical Trail Guidance Documents , and relevant ISO or EN standards. Additional documents included each month. Compliance with EN 46001 and 21 CFR 820 is essential for medical devices.
UK company MIOTIFY has developed a web-based software platform that product teams can use to configure medical algorithms quickly, create code packages to harness them and generate supporting documentation – all in a format that is designed to meet regulatory requirements for software as a medical device (SaMD). Sheena Macpherson.
The NDRP focused on six strategic objectives one of which was the implementation of the Integrated Assessment which per CDER was designed to “critically, collaboratively, and consistently assess whether information in drug approval applications meets legal and regulatory requirements.” 89 FR 74966 at 74968 (Sept.
The sampling strategy must be supported by sound and properly cited sources whose conclusions must be presented as supportive elements in the study documents. The following four elements of sampling methodology were found to be under-documented in RMM effectiveness studies: Supporting documentation for country/region selection.
Subscribe on iTunes , Android , or Stitcher If you navigate to the page on the website for the Institute for Safe Medication Practices that once hosted the document titled “Oral Dosage Forms That Should Not Be Crushed” you will instead find a notice that the list has been removed from the ISMP website.
In a budget change proposed in February and confirmed in May , California’s Department of Health Care Access and Information (HCAI) requested a one-time investment of $100 million for Newsom’s CalRx Biosimilar Insulin initiative. The state plans to work directly with a contract manufacturing organization (CMO) to manufacture low-cost insulin.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
The document contains detailed information on the suppliers and their product offerings, alongside contact details to aid your purchasing or hiring decision. Quality in terms of number of defective items, packaging and labelling, quality management system certification, research, development, and innovation.
Within the EU, patient information leaflets (PILs) are not merely a regulatory requirement but a cornerstone of patient safety. These standardised documents provide meticulously curated and scientifically approved information. 11 This requirement directly contradicts what is included in Article 63.
As a result, nutraceutical startups need to stay informed about the latest regulatory changes and requirements in the states where they do business. To achieve this, startups should implement rigorous quality control measures throughout their production processes, from sourcing ingredients to manufacturing and packaging.
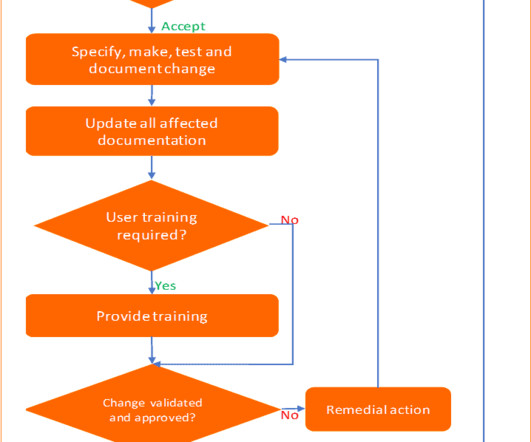
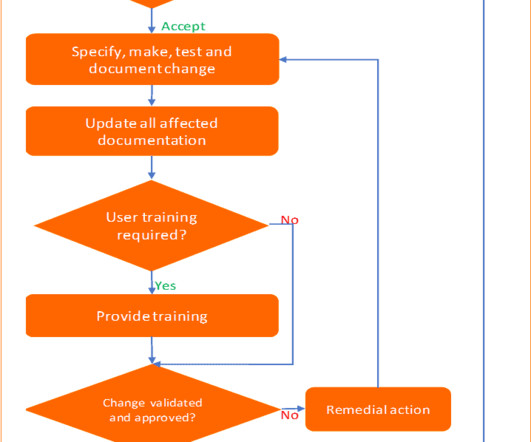
Imagine your company is going to install new information management software in the laboratory to replace the old one. The keywords in any change control management process are compliance with regulatory requirements, impact assessment, change verification, and maintain documentation. Additional documents included each month.
Imagine your company is going to install new information management software in the laboratory to replace the old one. The keywords in any change control management process are compliance with regulatory requirements, impact assessment, change verification, and maintaining documentation. Additional documents included each month.
Health Canada is seeking input from industry stakeholders on a new draft guidance document that discusses the use of electronic media in prescription drug labelling. Consultation on the draft guidance document is open until May 7, 2021. Any non-drug information must be derived from trusted, high-quality sources.
Line clearance ensures the processing line is free of any irrelevant products, components, and documentation that could be left accidentally from an earlier batch. Line opening ensures correct products, components, and documentation are brought into the processing line so the operation can commence without error.
Marketing Packages Taking part in the Excellence Awards offer your company many benefits including being able to tell the market, new and prospective clients about your achievement. For full details on the benefits, the scale of our audience and the marketing packages on offer please download the RESEARCH GUIDE.
and Europe held manufacturers accountable for their environmental impact, leading to an increase in partnerships with tech companies focused on sustainable packaging and production solutions. For patient-centric innovations to succeed, patients must be informed about their options and the benefits of personalised treatments.
The document contains detailed information on the suppliers and their product offerings, alongside contact details to aid your purchasing decision. Role of freeze-drying systems in the pharmaceutical industry The pharmaceutical freeze-drying market is highly governed by stringent standards and laws.
The exemption applies to “any … eligible trading partners, which … have successfully completed or made documented efforts to complete data connections with their immediate trading partners.” Know where your current product transaction information is being stored and how to access it.
It will also produce policy documents and informationpackages for patients, doctors and senior management in companies, Other workstreams could include computational strategies to make such simulations more powerful and efficient, new curricula to educate the workforce on the development and use of the technologies.
manufacturing, packaging, testing, storage and/or distribution of products) shall be validated to provide assurance that their system is capable of producing without error and that the results are robust and reproducible.” Additional documents included each month. 240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms.
“Thankfully, paper-based studies are becoming a thing of the past, but spreadsheets and Word documents are still commonly used to design and build trials. It can also lead to difficulties in finding information, as it is often stored in disparate systems each with their own access rights and permissions.”.
API DMFs are documents containing information on APIs that are submitted to the U.S. Food and Drug Administration (FDA) by API manufacturers to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products.
Cross contamination is dangerous in pharmaceutical industry as it can severely compromise patient safety if the content is not pure and the product package differs from what is written on the label. Additional documents included each month. What are the examples of cross contamination in pharmaceutical industry?
According to cGMP, validation in pharmaceutical industry can be defined as “Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes.” Additional documents included each month.
There are common, though preventable, pitfalls that applicants may encounter, including incomplete or inadequate nonclinical data, which can be addressed by ensuring that the package of nonclinical studies submitted in the application adhere to applicable ICH and FDA guidelines.
This amendment marks the first significant revision of Part 820 since 1996, which established the Quality System (QS) regulation and “included requirements related to the methods used in, and the facilities and controls used for, designing, manufacturing, packaging, labeling, storing, installing, and servicing of devices intended for human use.”
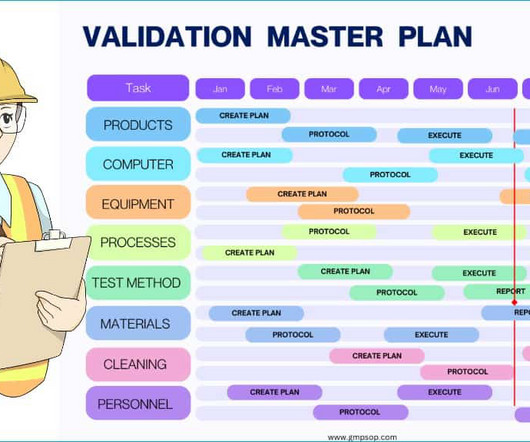
A validation master plan (VMP) is a strategic document that identifies the elements to validate, the approach to each element, organizational responsibilities, and a strategy for maintaining validation documentation. The VMP should provide information on: – Which items are subject to validation or qualification?
Acceptable Quality Limit is used to make an informed decision whether to accept or reject an incoming packaging components lot by assessing the lot size and types of defects found during a pre-determined level of inspection. To come to a methodical conclusion, the sampling inspector has to know the following information.
Additional documents included each month. If significant trends are identified, they should be documented, and changes should be made to avoid out-of-specification results. The number of batches manufactured, released and rejected should be documented. 240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms.
The information contained within the download document is intended for pharmaceutical manufacturers, wholesalers, retailers and distributors, pharmaceutical executives, medical representatives, business development managers, retail salesmen, sales managers, pharmacy executives, and any other individual involved in pharmaceutical marketing.
Such codes need to be placed on device labels and packages to allow devices to be easily identified and tracked throughout their lifecycle, except where the rule provided for an exception or alternative. In addition, the final rule requires the submission of product information to the Global Unique Device Identification Data (GUDID).
The Hidden Truth Studies have shown that the antineoplastic residues can be found be on the external packaging of both oral and IV drugs. Change gloves every 30 minutes during compounding or immediately when damaged or contaminated, unless otherwise recommended by the manufacturer's documentation.
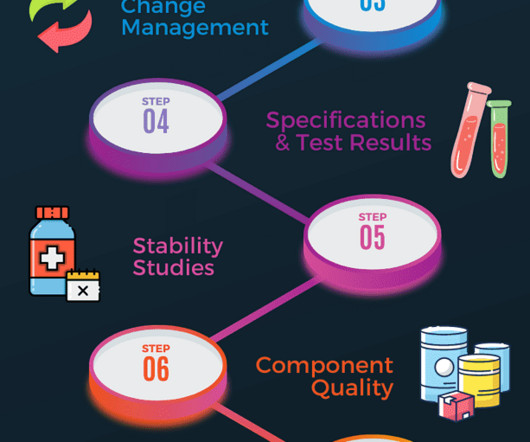
Stability testing ensures that the manufactured products remain safe, pure, and effective throughout their shelf life if they are kept in specified packaging and under environmental conditions. Additional documents included each month. Stability protocols are required to create a testing regime. All written and updated by GMP experts.
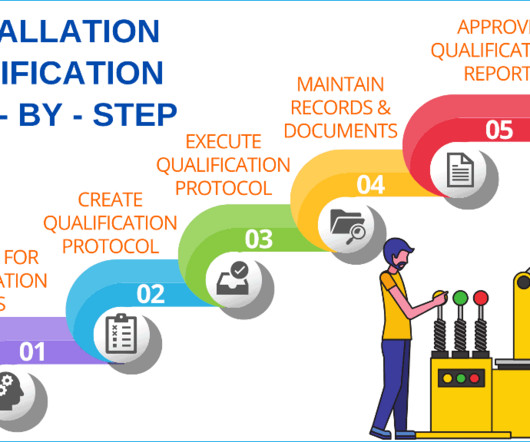
Installation qualification will provide you with documented evidence that the equipment or system has been designed, developed, supplied, and installed in accordance with design drawings, the supplier’s recommendations, and in-house user requirements. Additional documents included each month. Checkout sample preview s.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content