This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This document is revised from a version published in October 2023. Firstly, leachables can compromise a products quality and therapeutic effect, by potentially interacting with the formulated drug product, the document reported. Moreover, manufacturers should documentinformation about their safety thresholds, the guidance stated.
To confirm your test results are trustworthy and unbiased, you would turn to well-designed policies, procedures, guidelines, methods, protocols and all types of records. In other word, you need GMP documentation specially developed and used for your laboratory to guide you through everything you do in the lab.
A pharmaceutical manufacturing plant compliant with Good Manufacturing Practices must have a cleaning validation program in place to establish documented evidence that the cleaning processes will consistently meet expectations by removing the traces of residues from the earlier products. Additional documents included each month.
Additional documents included each month. employee assay qualification) must be documented in an SOP for each assay the lab performs with pre-determined acceptance criteria. employee assay qualification) must be documented in an SOP for each assay the lab performs with pre-determined acceptance criteria. gown qualification).
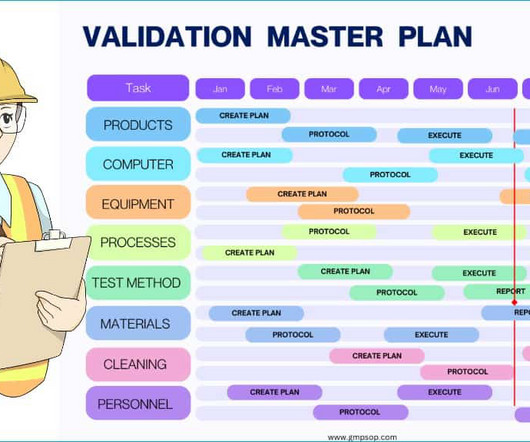
This article can help you understand who is responsible for preparing the validation master plan, the stages of the validation life cycle, the risk-based prioritization of validation items, how to prepare a validation schedule, and some practical examples. What is a validation master plan (VMP)? 12 or 24 months).
Table of Contents What is the concept of validation in pharmaceutical industry? ” Validation is a critical concept in the pharmaceutical industry. .” ” Validation is a critical concept in the pharmaceutical industry. Additional documents included each month. All written and updated by GMP experts.
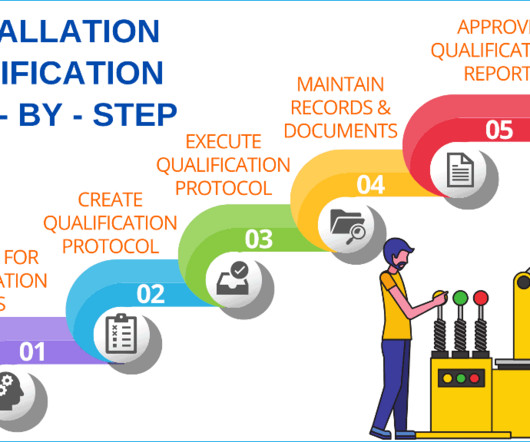
Installation qualification will provide you with documented evidence that the equipment or system has been designed, developed, supplied, and installed in accordance with design drawings, the supplier’s recommendations, and in-house user requirements. Additional documents included each month. Checkout sample preview s.
The analyst should ensure that the supervisor is notified as soon as an OOS event occurs, the sample is retained, and a documented investigation is commenced. – Document control records management and archiving systems. Additional documents included each month. All written and updated by GMP experts.
Additional documents included each month. The points below outline some areas in the codes where G(C)LP regulations are embedded in the GMP regulations and guidance documents. Additional documents included each month. All sampling procedures and plans must be documented. All written and updated by GMP experts.
Additional documents included each month. If significant trends are identified, they should be documented, and changes should be made to avoid out-of-specification results. The number of batches manufactured, released and rejected should be documented. Additional documents included each month. Checkout sample preview s.
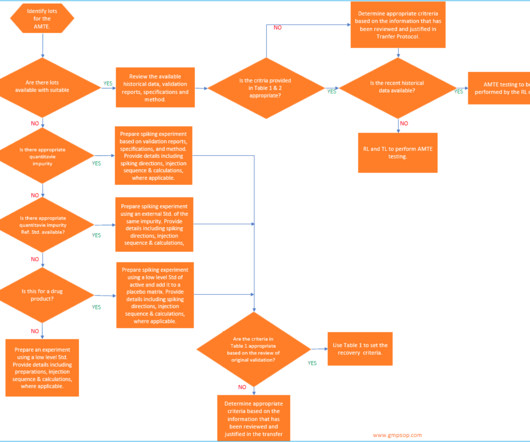
According to USP <1224> , method transfer is defined as the documented process that qualifies a laboratory (RL) to use an analytical method originating from another laboratory (TL), regardless of whether it’s internal or external. Additional documents included each month. Checkout sample preview s.
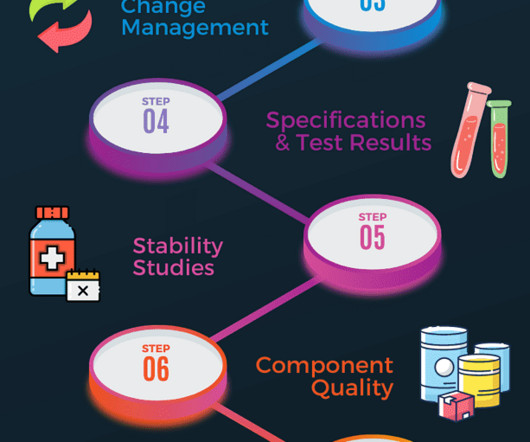
It should have the minimum information, such as general product information, specifications and test methodology, results, data analysis and estimated shelf life. Additional documents included each month. They are necessary for the API testing method and will also have to be conducted for the formulated and packaged product.
Additional documents included each month. Quality management system is primarily involved in ensuring good manufacturing practice (cGMP), good laboratory practice (GLP), good documentation practice (GDP), good automated manufacturing practice (GAMP) etc. What is the probability my cleaning methods weren’t validated?
The guidance encourages sponsors to engage with FDA using the Q-Submission Program prior to submitting a PCCP in order to obtain FDA feedback on if the proposed modification is suitable for inclusion in a PCCP and what information the PCCP will need to include. FDA may request additional information during the review of the PCCP.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

















Let's personalize your content