This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. Note that section 3060(a) of the 21st Century Cures Act in 2016 amended section 520 of the FD&C Act and removed certain software functions from the statutory definition of a medical device. Loose Ends IDEs.
Both ISO 13485 and ISO 9000 contain terms and definitions that are referenced within Part 820. includes considerations with respect to documentation, applicable regulatory requirements, design and development, and enforcement. FDA further retained some definitions in the QSMR. Certain definitions are also removed from § 820.3,
The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). The FDA regulates software that meets the definition of a medical device in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act). Criteria for regulation.
States frequently review labels (and labeling) for animal food products. A major component of AAFCO is its work on ingredient definitions, specifying what ingredients may be use in animal feed under what conditions. AAFCO’s ingredient definitions are not federal regulations and do not have the force or effect of federal law.
In October 2023, the European Parliament (EP) proposed revisions to the European Commission proposals with diverging views on various topics set out through two documents. In October 2023, the European Parliament (EP) proposed revisions to the European Commission proposals with diverging views on various topics set out through two documents.
A pharmaceutical warehouse is responsible for receiving, storing, and releasing incoming goods (including labeling and packaging) and distributing finished products. Therefore, GMP rules ensure that materials are handled and stored properly and that appropriate documentation is maintained. Additional documents included each month.
Additional documents included each month. Packaging/labeling/Misbranding: 41 warnings vi. Similarly, the packaging, labeling, and misbranding-related warning letters were the top reasons in 2019. Additional documents included each month. Additional documents included each month. Checkout sample preview s.
However, as with drugs, dietary supplements require evidence that they are safe and that claims on product labels are truthful and not misleading. It begs the question of why only two botanical drug products have obtained FDA approval and fulfilled the Botanical Guidance definition of a botanical product.
Assessing the impact of changes to a process/system The use of quality risk management and the methodology to be used should be documented in the relevant validation plan or change management procedure. Additional documents included each month. process change, product recall, labelling change, adverse event/safety reporting)?
These standards can only be used if they are standardized against primary standards using definitive methods that are published in pharmacopoeias, and national and international standards. Additional documents included each month. d) When the reference standard is first opened, an “opened date” must be written on the label of the vial.
SODA - Sale of Drugs Act 1952 CDCR - Control of Drugs and Cosmetics Regulations 1984 DCA - Drug Control Authority Key Definitions "Drug" includes any substance, product or article intended to be used or capable, or purported or claimed to be capable, of being used on humans or any animal, whether internally or externally, for a medicinal purpose.
Introduction Once, I received a query from a ward nurse: If a product label mentioned that "Do not store over 25°C", is it fine for us to keep the medication in fridge? What does a cool place on the product label mean? Read the label to know the recommended storage temperature range.
By definition, corrective actions are required to eliminate the causes of an existing nonconformity, defect, or other undesirable situation. Additional documents included each month. Additional documents included each month. Corrective actions are taken to fix a defect on hand. All written and updated by GMP experts.
Serial testing was defined as testing symptomatic individuals twice over three days with at least 48 hours between tests, which is in accordance with study findings from the National Institute of Health referenced here and current EUA labeling for serial testing. Customer Support Help line).
In 1997, the Food and Drug Administration Modernization Act (FDAMA) amended the definition to make clear that substantial evidence could also be demonstrated by “one adequate and well-controlled clinical investigation and confirmatory evidence.” However, the Act does not define what may constitute such confirmatory evidence.
CBER may still request additional information when deemed appropriate, but the stated hope is that increased use of VCS can facilitate product development by reducing the need to develop unique methods for individual products and that they will typically reduce the amount of necessary documentation “and may reduce FDA review time.”
Additional documents included each month. Quality management system is primarily involved in ensuring good manufacturing practice (cGMP), good laboratory practice (GLP), good documentation practice (GDP), good automated manufacturing practice (GAMP) etc. The quality risk management framework is ingrained in its definition.
These documents focused on why the sentinel event occurred. Both the label and the product had to match the order before the product. No, but something was definitely wrong when I was checking the IV solutions.”. We found nothing wrong with the IV solutions or their labeling. Blaming Employees For Sentinel Events.
The NHC requests more clarity on how CMS will exclude QALY-based metrics and highlight when they have been removed from consideration in MFP justification documentation. Without appropriate guardrails, CMS’ broad definition of drugs eligible for negotiation may discourage these types of improvements. hstc=117268889.c6acac5669d4f1e6063a774e6d96c6b5.1716560813145.1716560813145.1716560813145.1&
Medical affairs definition uses clinical and scientific information to communicate the efficiency of a drug. They provide information about the following: Off-label usage Publications Safety information Independent medical education. What do medical affairs teams do? .
For example, CMS has implemented CED for novel amyloid beta-targeting therapies for Alzheimer’s disease even when these therapies are being used according to their FDA- approved label and accepted medical practice, and continued CED even once products have transitioned from accelerated approval to traditional approval.
What are the legal requirements of labelling on a medication for a dispensed medicine by pharmacist? What is the definition of Zero and first order reaction Bioavailability Elimination rate constant Half-life Steady-state Given a drug has volume distribution of 0.2 How long should a fully dispensed prescription be kept for?
Terazosin - Off-label use in ureteral stone(s) expulsion Topical Corticosteroids - Apply thinly on the skin using fingertip unit. Statins - Not all statins need to be taken at night.
Gaulkin & Ritte van Laack Readers of this blog likely are familiar with the ongoing, often contentious battle over the labeling of plant-based foods (see, e.g., here , here , and here ). For foods that do not have established definitions and standards of identity (i.e., The description or name of the food (i.e.,
A few years ago, I had a few clients with documented elevations in prolactin, and I have seen that lowering prolactin levels can help with reducing thyroid antibodies. Studies have, however, documented that bromocriptine, a medication used to lower prolactin levels, can reduce flares in lupus, another autoimmune condition.
The proposed revised rule constituted a radical change from the original definition of healthy, which focused on the presence of individual (beneficial) nutrients. Under the proposed rule, any raw whole fruits and vegetables could be labeled healthy no matter the level of added sugars, sodium, or saturated fat.
The manufacturer should, in addition to stating whether the modification will be implemented manually or automatically, include details such as: End user actions needed, if any to implement the change, Timing of implementation, Extent of implementation in the install base, and Include references to expected labeling changes.
Upon receipt of the 513(g), FDA will assess whether the product meets the definition of a device under section 201(h) of the FD&C Act. Provided that a brief device description, clear intended use, and list or picture of all labeling claims are made, FDA aims to review the information and provide a response generally within 7 days.
To all external observers, the medication looked legitimate – it bore a normal label, it was the right size, the product tracing documentation was received in good order. Biktarvy® and other HIV drugs are generally labeled to “dispense in original packaging” so the pharmacist never broke the seal on the bottle.
Lenz, Principal Medical Device Regulation Expert In early January, FDA released a draft guidance document titled Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations (Draft AI Guidance). By Adrienne R. User Interface Software Description Section VI.B
Fermented foods are also great, and are definitely one of my 12 favorite beneficial healing foods for the gut. As such, I re-analyzed the data for the “presence” of parasite DNA labels, and the incidence was substantially higher… 274 total parasites were detected! (See my article on probiotics for more information on this.)
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.







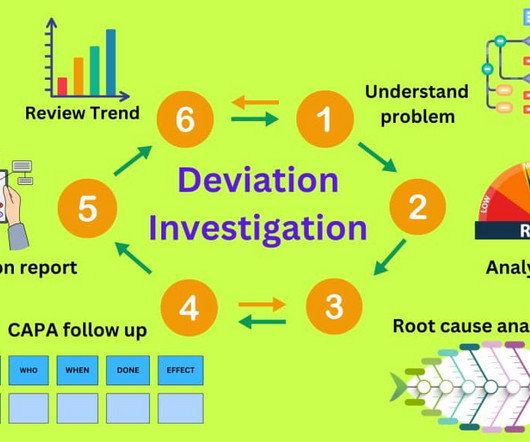

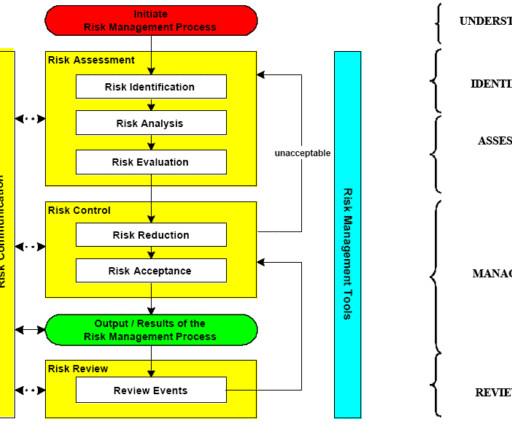



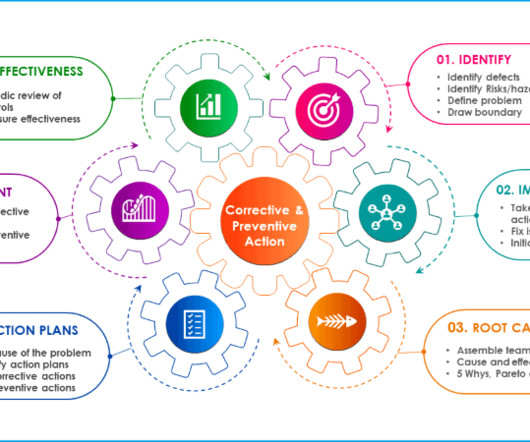
























Let's personalize your content