This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In general, you must formally validate all process steps, production equipment, cleaning methods, testing methods, computer systems and environment, facilities and utilities which are directly used for the manufacture of sterile and non-sterile products. This validation is relatively less comprehensive.
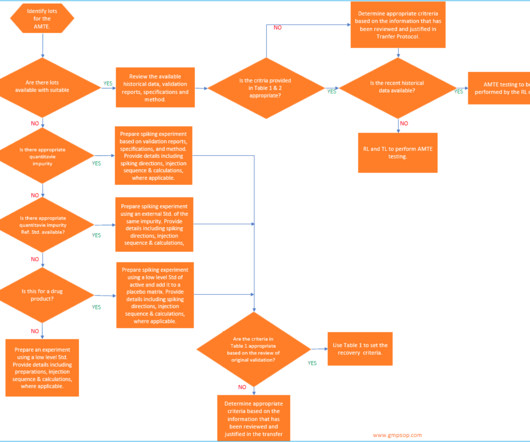
According to USP <1224> , method transfer is defined as the documented process that qualifies a laboratory (RL) to use an analytical method originating from another laboratory (TL), regardless of whether it’s internal or external. What is the analytical method transfer protocol?
According to FDA Guidance for Industry – Q9 Quality Risk Management , “Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the drug product across the product lifecycle. If my cleaning methods are not validated would they cause contamination of my products?
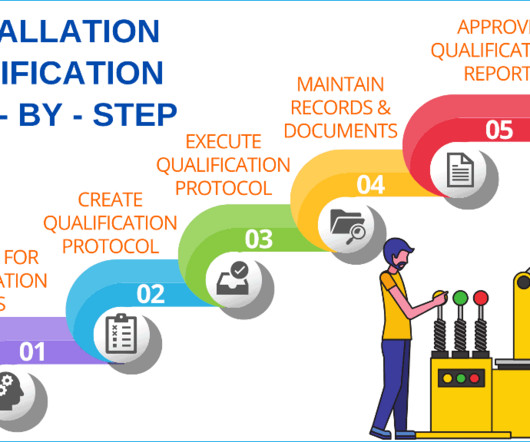
Add the objective of the installation qualification protocol Write the objective of the protocol defining the installation qualification (IQ) and operational qualification (OQ) requirements and acceptance criteria for the equipment with location i.e., packaging or manufacturing, and the facility.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









Let's personalize your content