This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
GSK skinny label case , the U.S. Specifically, the Government explained, “[t]he section viii pathway cannot function properly if FDA and generic manufacturers cannot rely on an NDA holder’s representations to the agency regarding which portions of the brand-name drug’s labeling teach patented methods of use.”
The updated guidance includes information on the general requirements for the content of FDA-regulated promotional communications about reference or biosimilar products and provides some examples to illustrate the FDA’s current recommendations for addressing reference, biosimilar, and interchangeable biosimilar products in product promotion.
“Where it is impossible to restrict access to HCPs, due to the congress platform or equivalent, there must be a clear statement to the attendee that the materials/communication are designed and intended for HCPs only.”. Here, we take a look at the top five takeaways from the document: 1. Identifying the appropriate code and label.
Apply Data: Regulatory employees should be using stored data to intelligently create submission documents. By limiting documents full of free text fields and subjectivity companies can adopt a more digitised approach, where document templates can be compiled automatically from available data. Label Authoring and Tracking.
These include Meditag serialised hologram labels, first introduced in 2005 and found on all registered medicines in Malaysia to help pharmacists and inspectors detect unauthorised and counterfeit product. Elsewhere, holographic labels have been deployed effectively to arrest declining sales of pharmaceuticals.
There are two means of gaining insight into the agency’s thinking about regulatory issues related to promotional communications by pharmaceutical companies; one is through the issuance of guidance documents, the other is through enforcement. But when it comes to enforcement things have changed greatly over the years.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The technological characteristics in this context may cover a wide range of device functions, for instance, monitoring features, stimulation parameters, and communications with healthcare providers. Loose Ends IDEs.
As noted in the past, there are two primary means for understanding the agency’s latest thinking with respect to promotional communications from pharmaceutical companies – the content and pattern of enforcement and the issuance of guidance documents to shed light on the parameters that exist.
Section IV, Additional Resources, provides links to previously-issued guidance documents and other educational materials geared to traditional device manufacturers, with no additional commentary on how to apply these requirements to the very different clinical laboratory environment.
FDA also requests a “detailed description of the allegation with any available supporting documentation.” In our experience, the more detailed the documentation that accompanies the submission, the more likely FDA will follow-up on the submission. There are drawbacks to the current communication channels: 1.)
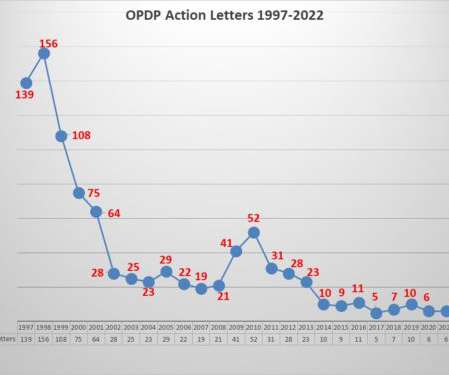
In the now distant past, enforcement from this office, then called the Division of Drug Marketing, Advertising, and Communications (DDMAC) and later re-named OPDP, was robust, with the office issuing scores of letters a year (156 were issued in 1998). One of them was about the appropriate use of links.
billion for the illegal marketing of four of its off-label drugs, which became one of the biggest fraud settlements in the healthcare industry. Just a small error could lead to many people adhering to compliance documentation and making the same mistake in their work. For example, Pfizer was fined $2.3
FDA communicates via a Substantive Interaction to inform the submitter either that FDA will proceed with Interactive Review or that the 510(k) will be placed on hold until FDA receives a complete response to an Additional Information request. When referencing documents within the AI response, cite location within the 510(k) supplement (e.g.,
The huge rise in the use of telemedicine services during lockdown has brought care directly to patients in their homes, while pharma’s communications with healthcare professionals (HCPs) has experienced a similar push to digital channels. The mindset that accomplished this needs to be retained,” Jeremy says. About the interviewees.
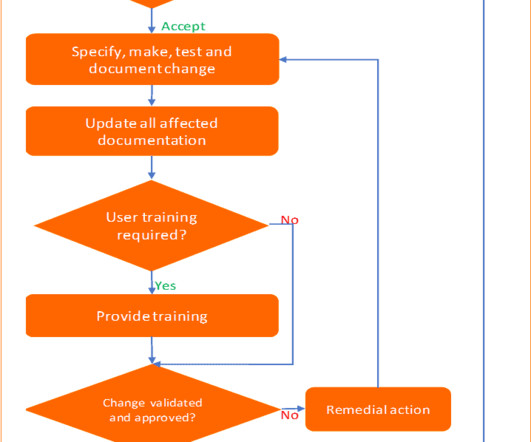
Change control management is a systematic process by which a change to facilities, products, systems, or processes is proposed, assessed by a committee (technical and operational impacts), approved, implement, reviewed for effectiveness, and communicated to a larger audience. All documentation must be kept as supporting evidence.
Change control management is a systematic process by which a change to facilities, products, systems, or processes is proposed, assessed by a committee (technical and operational impacts), approved, implement, reviewed effectiveness, and communicated to a larger audience. All documentation must be kept as supporting evidence.
Medical affairs in Pharma are often seen as a central agency that works within a healthcare company and prioritize communication among life science organizations, medical professionals, healthcare providers, and patients. Medical affairs definition uses clinical and scientific information to communicate the efficiency of a drug.
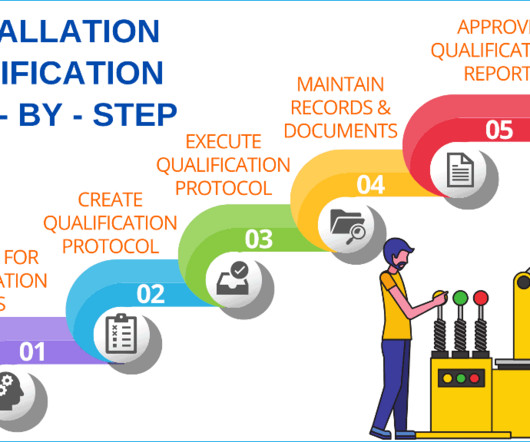
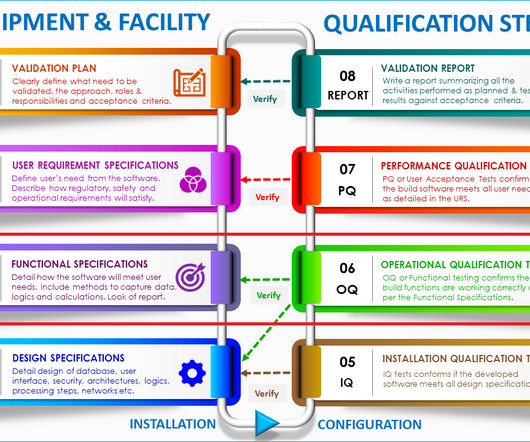
Installation qualification will provide you with documented evidence that the equipment or system has been designed, developed, supplied, and installed in accordance with design drawings, the supplier’s recommendations, and in-house user requirements. Additional documents included each month. Checkout sample preview s.
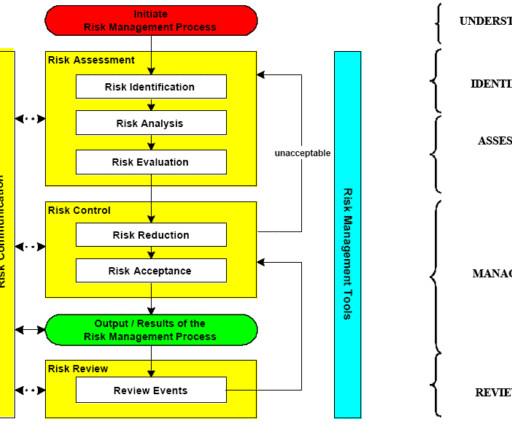
Assessing the impact of changes to a process/system The use of quality risk management and the methodology to be used should be documented in the relevant validation plan or change management procedure. Additional documents included each month. process change, product recall, labelling change, adverse event/safety reporting)?
This technology is reforming the management of prescriptions, enhancing the accuracy of medication dispensing, streamlining communication with healthcare providers, and allowing pharmacists to devote more time to patient care. In traditional settings, communicating such critical information can take hours or even days.
But it is not likely to surprise anyone that the first half of 2020 has caused not only an increase in communication from FDA, but a focus as well. The agency sent out 9 alerts during the first half of the year, 4 of which were related to COVID-19, including one for the off-label use of anti-malaria medications.
Raw materials must be inspected to confirm that the containers are intact, have been provided according to the paperwork, and have labels affixed on them identifying the raw material name, batch number, and expiry date. For example, materials with Hold, Quarantine, or Rejected labels must be kept in a quarantine location.
The FDA explains that regardless of the complexity of the software and whether or not the software is proprietary, the output or labelling should provide HCP users with adequate background information in plain language on the input(s), algorithm logic or methods, datasets, and validation. Criteria for regulation.
All waste generated during the cleanup should be disposed of properly in labeled bags, and the equipment used must be cleaned and stored appropriately. Additional documents included each month. Examples include oil leaks from equipment in a packaging line or small solvent spills in the printing and labeling machine.
By Philip Won & Véronique Li, Senior Medical Device Regulation Expert — As we recently blogged , FDA released three draft guidance documents to help enhance the predictability, consistency, and transparency of the 510(k) program. One of these documents focuses on “ Evidentiary Expectations for 510(k) Implant Devices.”
Additional documents included each month. After the preparation secondary reference standards are tested for potency, labelled, dated and stored, handled and used in testing so as not prejudice quality. d) When the reference standard is first opened, an “opened date” must be written on the label of the vial.
Additional documents included each month. Additional documents included each month. Cleaning requirements for dedicated equipment may be less than those for common equipment, provided the company has documented either the maximum number of batches or the maximum time that can elapse before complete cleaning must be done.
Additional documents included each month. Quality management system is primarily involved in ensuring good manufacturing practice (cGMP), good laboratory practice (GLP), good documentation practice (GDP), good automated manufacturing practice (GAMP) etc. Additional documents included each month. Checkout sample preview s.
Manufacturer-Focused Input Question 30: Off-Label Use. CMS has appropriately highlighted the significance of off- label use information, providing a specific avenue for manufacturers to submit data on off-label uses supported by evidence-based guidelines listed in CMS-recognized Part D compendia.
Immediately communicate the incident to stakeholders and initiate an internal laboratory investigation. Additional documents included each month. – Status control: implementing status labels to clearly identify workflow status such as issues, returns, rejects and reworks of components to prevent mix ups.
ampules, bottles, labels, cartons, shipping containers, desiccants) Services (e.g., This risk assessment effort is not a one-time event but rather a periodic, recurring process for communicating and reviewing risks,” noted USP’s Horacio Pappa, Ph.D., active ingredients, excipients, other raw materials) Packaging materials (e.g.,
CBER may still request additional information when deemed appropriate, but the stated hope is that increased use of VCS can facilitate product development by reducing the need to develop unique methods for individual products and that they will typically reduce the amount of necessary documentation “and may reduce FDA review time.”
As noted in our previous communications, while the NHC would prefer a more traditional Notice and Comment rulemaking opportunity that would ensure the Agency directly responds to stakeholder feedback, we welcome this opportunity to express our reactions to CMS’ thinking on the negotiation program.
Additional documents included each month. Additional documents included each month. You should identify and document the instrument ID, date and time of calibration, units of measurement, the standards used, the steps taken during calibration, and environmental conditions. Additional documents included each month.
To maintain the integrity and safety of these investigational products, you must adhere to Good Manufacturing Practice (GMP), FDA Clinical Trail Guidance Documents , and relevant ISO or EN standards. Additional documents included each month. Additional documents included each month. ensure traceability and compliance.
These requirements mandate that pharmaceutical companies thoroughly document OQ protocols, testing methods, acceptance criteria, and results. Installation qualification verifies and documents that all critical aspects of equipment installation adhere to predetermined specifications and manufacturer’s recommendations.
The overarching principle set out in Codes of Practice, and in particular the Principles for the use of digital channels in the EFPIA Code , is that the legislation and Codes of Practice apply equally to communications by companies on social media and digital channels.
Terazosin - Off-label use in ureteral stone(s) expulsion Topical Corticosteroids - Apply thinly on the skin using fingertip unit. Statins - Not all statins need to be taken at night. Human Touch - Why Artificial Intelligence cannot replace us still? Theory in Practice - Life is not ideal but a struggle Life - Why?
Procedures for Submitting the DAP Sponsors should describe the DAP clearly and concisely, with limited cross-referencing to previously submitted documents. If FDA determines that a waiver will be issued, it may consider public communications about the decision. Would any such failure to meet DAP goals be reflected in labeling?
Please consider the following and be sure to read the precautions on individual labels before you try a new herb, supplement, or blend. (In Holy Basil ( Ocimum sanctum ) The anti-inflammatory effects of holy basil (also known as tulsi) have been documented in many in vitro and in vivo studies. Exp Toxicol Pathol. Exp Toxicol Pathol.
Issues with regulatory requirements and documentation can also cause significant delays, while any inaccuracies in translations on labelling can mean that dosage and storage information is not correctly understood or followed. And accurate labelling and translation are critical for this sector.
A few years ago, I had a few clients with documented elevations in prolactin, and I have seen that lowering prolactin levels can help with reducing thyroid antibodies. Studies have, however, documented that bromocriptine, a medication used to lower prolactin levels, can reduce flares in lupus, another autoimmune condition.
The manufacturer should, in addition to stating whether the modification will be implemented manually or automatically, include details such as: End user actions needed, if any to implement the change, Timing of implementation, Extent of implementation in the install base, and Include references to expected labeling changes.
According to the legal documents , the plaintiffs accused the companies of “violating federal antitrust laws, alleging “per se” and “rule of reason” violations”. Novartis argued that the early release of Entresto generics could cause labelling inconsistencies. Please check your email to download the Report.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content