This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The International Organization for Standardization (ISO) has published its new standard Sterilization of health care products — Microbiological methods — Part 3 Bacterial endotoxin testing ( ISO 11737-3:2023). The document contains requirements and guidance for testing for bacterial endotoxins.
Draft guidance published by the US Food and Drug Administration (FDA) in December 2023, discussed quality considerations for topical ophthalmic drug products, including key considerations for extractables and leachables (E&L) testing. This document is revised from a version published in October 2023.
In other word, you need GMP documentation specially developed and used for your laboratory to guide you through everything you do in the lab. Depending on the objective of the lab, the need for GMP documentation will change for the laboratory. The significance of well-designed GMP documentations is immense.
Operators On 28th Jan’ 2023 Job Description We are hiring for the following vacancies in RPL On urgent Basis. Operators On 28th Jan’ 2023 Rakshit Pharmaceuticals Ltd- Walk-In Interviews for Production-Jr. Department: Production Position: Jr. Operators Experience: 0 years Qualification: S.S.C/
This provision became effective as of March 29, 2023. It will become part of the “refuse to accept” (RTA) checklist on October 1, 2023. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. Timeline Section 524B became effective on March 29, 2023.
Such codes need to be placed on device labels and packages to allow devices to be easily identified and tracked throughout their lifecycle, except where the rule provided for an exception or alternative. The compliance dates were first published in 2013, and subsequently updated in various guidance documents and regulations published by FDA.
We now see that the proposed rule to “harmonize and modernize” the QSR with ISO13485:2016, creating the new QMSR, is on the Spring 2023 Unified Agenda (see here ). In fact, the priority designation for the final rule is labeled as “economically significant.” According to the Unified Agenda, the proposed rule is in the final rule stage.
The regulator has set 29 August 2023 as a Prescription Drug User Fee Act (PDUFA) goal date. Outlook Therapeutics president and CEO Russell Trenary said: “This BLA acceptance and PDUFA date are significant milestones in our mission to offer clinicians and their patients the first and only on-label, ophthalmic bevacizumab to treat wet AMD.
Consultation on the Regulatory Amendments, which have been published in the Canada Gazette, Part 1 , is open until March 27, 2023. Feedback on the Regulatory Amendments and draft guidance documents can be submitted to Health Canada here.
The long-awaited final rule, which we last discussed in a July 2023 blog post and have tracked in our March 2023 and March 2022 posts, aims to harmonize quality management system requirements for medical devices with requirements set forth by other regulatory authorities around the world. Notably, Part 820 will look different.
On June 12, 2023, FDA issued a public notice to solicit comments on the information collection related to the voluntary submission of allegations of regulatory misconduct to CDRH. Any comments to the public notice must be submitted by August 11, 2023.
In Q1 2023, the European Commission (EC) announced proposed changes to EU pharmaceutical legislation. In October 2023, the European Parliament (EP) proposed revisions to the European Commission proposals with diverging views on various topics set out through two documents. Internet] 2023. Internet] 2023.
requesting data and documents regarding their business and business practices. In 2023, the FTC issued supplemental Orders to three additional PBM-affiliated entities. pharmacies in 2023, demonstrating this horizontal consolidation. OptumRx, Inc.; Humana Pharmacy Solutions, Inc.; billion prescriptions dispensed by U.S.
This is the third withdrawal of an accelerated approval FDA has performed, following Avastin in 2011 and Makena in 2023. In contrast , the two previous withdrawals, Makena in 2023 and Avastin in 2011, took 30 months and 11 months, respectively. On August 4, 2023, Oncopeptides submitted its appeal.
a) , and related guidance documents (e.g., Post-market, manufacturers can make modifications consistent with the PCCP and document the modification in accordance with their quality system, without the need for a new marketing submission. See 21 CFR 807.81(a)(3) a)(3) and 21 CFR 814.39(a)
The TLR9 agonist is currently being investigated in the Phase III CONCLUDE trial (NCT04985968), with an expected study completion date of August 2023. This is an important population to target, as it is well-documented that a number of patients are intolerant or non-responsive to first-line biologics like infliximab or adalimumab.
This randomised, open label-controlled multicentre clinical trial was conducted across 21 sites in Spain from 2021 to 2023. European Society of Clinical Microbiology and Infectious Diseases: 2021 update on the treatment guidance document for Clostridioides difficile infection in adults. Clin Microbiol Infect.
Packaging and labelling mix-ups can potentially result in serious health consequences. Accurate labelling of medicines is critical to patient health. Therefore, manufacturers pay particular attention to getting the right medication and strength into the right container with the correct labels and instructions for use.
States frequently review labels (and labeling) for animal food products. The CFI includes a list of common foods that “may be appropriate for use in animal food and serve as a tool for use during review of ingredients on an animal food label.” This document also describes an appeal procedure.
The GMP provisions require the EU and UK to recognise the other’s manufacturing facility inspections and accept its GMP documents. The sizeable gap in the TCA is the EU and UK’s failure to agree on mutual recognition of conformity assessments, approval bodies, product markings or labelling, other than the very limited provisions on GMP.
BCPS, BCIDP Article Posted 12 March 2023 As a pharmacist serving as a preceptor to students and residents in the area of infectious diseases since 2010, I have always been on the hunt for good infectious diseases journal articles for my learners to read. Authored by: Timothy P. Gauthier, Pharm.D.,
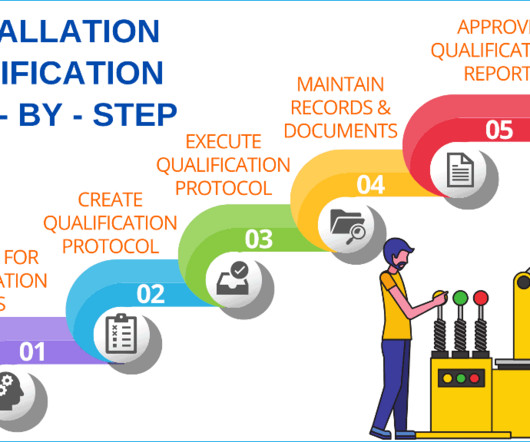
Installation qualification will provide you with documented evidence that the equipment or system has been designed, developed, supplied, and installed in accordance with design drawings, the supplier’s recommendations, and in-house user requirements. Additional documents included each month. Checkout sample preview s.
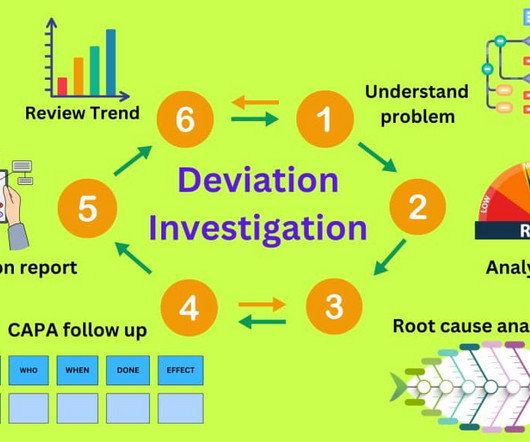
Additional documents included each month. In the five years between 2019 and 2023, the FDA issued the following warning letters, closely related to ineffective deviation investigation. Packaging/labeling/Misbranding: 41 warnings vi. Additional documents included each month. All written and updated by GMP experts.
Maybe that’s also why FDA last week publicized the highest number of important Warning Letters of the year (compared with prior releases in 2023). Perhaps FDA wanted us to remember 2023 as the year FDA succeeded in uncovering critical defects in drug and device manufacturing, and in critical trials. Terragene S.A., Terragene S.A.,
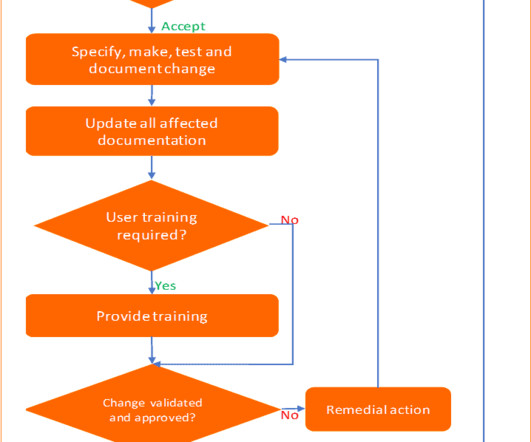
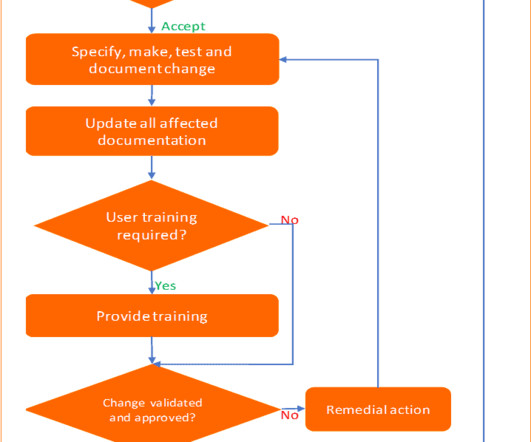
Six steps process to implement change control management Pharmaceuticals quality assurance & validation procedures GMPSOP Six steps process to implement change control management Last modified: June 3, 2023 Table of Contents “It is not the strongest of the species that survive, nor the most intelligent, but the one most responsive to change”.
Six steps process to implement change control management Pharmaceuticals quality assurance & validation procedures GMPSOP Six steps process to implement change control management Last modified: May 28, 2023 Table of Contents “It is not the strongest of the species that survive, nor the most intelligent, but the one most responsive to change”.
April 04, 2023: “Pfizer announced that the U.S. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by the FDA is in Fourth-Quarter 2023 for the sNDAs. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by the FDA is in Fourth-Quarter 2023 for the sNDAs. In the U.S.,
Mullen — As of October 1, 2023, all 510(k) submissions, unless exempted, must be submitted to FDA using the electronic Submission Template And Resource ( eSTAR ). On September 29, 2023, FDA released a draft guidance on Electronic Submission Template for Medical Device De Novo Requests. By Philip Won & Allyson B.
The analyst should ensure that the supervisor is notified as soon as an OOS event occurs, the sample is retained, and a documented investigation is commenced. – Document control records management and archiving systems. Additional documents included each month. All written and updated by GMP experts.
By Philip Won & Véronique Li, Senior Medical Device Regulation Expert — As we recently blogged , FDA released three draft guidance documents to help enhance the predictability, consistency, and transparency of the 510(k) program. One of these documents focuses on “ Evidentiary Expectations for 510(k) Implant Devices.”
Additional documents included each month. Additional documents included each month. Cleaning requirements for dedicated equipment may be less than those for common equipment, provided the company has documented either the maximum number of batches or the maximum time that can elapse before complete cleaning must be done.
Serial testing was defined as testing symptomatic individuals twice over three days with at least 48 hours between tests, which is in accordance with study findings from the National Institute of Health referenced here and current EUA labeling for serial testing. Customer Support Help line).
The new guidance is one of three policy documents dedicated to explaining FDA’s interpretation of this statutory authority and their approach to exercising scientific judgment in evaluating drug effectiveness. We are heartened to see that this latest guidance reflects many of the advances we observed in practice since 2019.
ampules, bottles, labels, cartons, shipping containers, desiccants) Services (e.g., josh.levin@usp.org Thu, 08/03/2023 - 15:43 Supply Chain active ingredients, excipients, other raw materials) Packaging materials (e.g., This builds public trust in medicines, foods, and dietary supplements.
However, as with drugs, dietary supplements require evidence that they are safe and that claims on product labels are truthful and not misleading. 1 For use as prescription drugs, a botanical product must be approved by the FDA: to date, only two have gained this approval.
Manufacturer-Focused Input Question 30: Off-Label Use. CMS has appropriately highlighted the significance of off- label use information, providing a specific avenue for manufacturers to submit data on off-label uses supported by evidence-based guidelines listed in CMS-recognized Part D compendia. and Keating, N.
The discussion focused on CMS’ 2023 listening sessions during implementation of the first round of negotiations and identified lessons learned to inform future listening sessions and broader patient engagement strategies. It is crucial to consider how off-label uses will be evaluated and incorporated into the negotiation process.
The primary endpoint evaluated in this study is progression-free survival, defined as time from randomization to the first documentation of progressive disease or death due to any cause, whichever occurs first. and Europe.” Key secondary endpoints include overall response rate and overall survival.
Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.” Tobolowsky & Richard A. It finalized a draft guidance published in 2022.
No surprise: Other manufacturers have followed Novartis’s lead As we reported in another previous post , on February 15, 2023 Johnson & Johnson took Novartis’s 40-mile restriction one step further. Fortunately, those steps tend to be the least burdensome in the process.
For example, CMS has implemented CED for novel amyloid beta-targeting therapies for Alzheimer’s disease even when these therapies are being used according to their FDA- approved label and accepted medical practice, and continued CED even once products have transitioned from accelerated approval to traditional approval.
Individual doctors may still be able to utilize this therapy with their patients as an “off-label” use. Remission of Hashimoto’s thyroiditis in a twelve-year-old girl with thyroid changes documented by ultrasonography. Accessed May 25, 2023. Accessed June 9, 2023. References [1] Nanan R, Wall JR. doi:10.1089/thy.2010.0102;
From the post: The HHS Budget in Brief document describes the policy objective simply enough: “ Permit Biosimilar Substitution without Prior FDA Determination of Interchangeability” and clarifies that this means “deem all approved biosimilars to be interchangeable with their respective reference products”.
The deadline for the Draft Guidance was December 29, 2023, so the draft, issued on June 26, 2024, is about 6 months late under FDORA’s mandate. Procedures for Submitting the DAP Sponsors should describe the DAP clearly and concisely, with limited cross-referencing to previously submitted documents.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content