This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
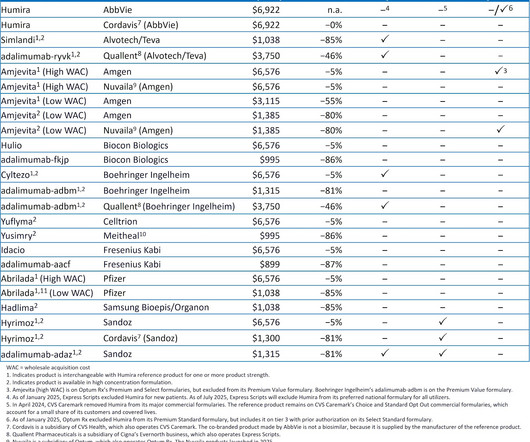
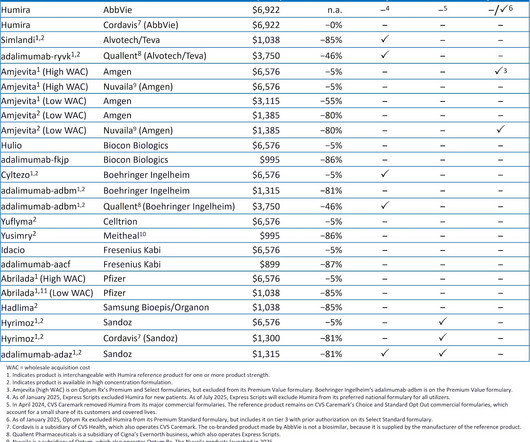
As youll see below, the combination of formulary exclusion and private labels is creating an increasingly confusing and crowded biosimilar marketplace. For 2025, the Big Three PBMs shifted national formularies to favor their private-labelbiosimilars over Humira and its many biosimilar competitors.
As youll see below, the combination of formulary exclusion and private labels is creating an increasingly confusing and crowded biosimilar marketplace. For 2025, the Big Three PBMs shifted national formularies to favor their private-labelbiosimilars over Humira and its many biosimilar competitors.
On October 30, 2024, Shanghai Henlius Biotech and Organon announced that the FDA has accepted the Biologic License Application (BLA) for HLX14, a proposed biosimilar to PROLIA/XGEVA (denosumab). In 2022, Shanghai Henlius Biotech granted Organon exclusive commercialization rights for HLX14 in the U.S., EU, and Canada. EU, and China.
Samsung Bioepis and Biogen have claimed the first FDA approval for a biosimilar version of Roche and Novartis’ Lucentis (ranibizumab) for leading causes of blindness, raising the prospect of a cheaper treatment option for US patients. The post FDA OKs first biosimilar of Roche’s blockbuster AMD drug Lucentis appeared first on.
This Revised Draft Guidance provides considerations for manufacturers, packers or distributors (dubbed “firms”) of prescription biological reference products, biosimilar products, and interchangeable biosimilar products presenting data and information about such products in promotional materials in a truthful and non-misleading way.
prescribers have high confidence in the safety and efficacy of biosimilars, a majority (58%) oppose third-party switching of a patient’s biologic medicine for non-medical (e.g. state law, only biosimilars which are interchangeable may be substituted by a pharmacist without contacting the prescriber. and worldwide.”
Our team at PharmaShots has summarized 15 key events of the biosimilar space of April 2023 1. Additonally, Rezvoglar was also approved in the US on Nov 2022 In addition to Rezvoglar, Lilly is the owner of Humulin (recombinant human insulin), Humalog (reference insulin lispro), as well as an unbranded insulin lispro product 2.
In preclinical studies, HLX15 is highly similar to daratumumab Amgen Launches First Humira (adalimumab) Biosimilar Amjevita in the US Date- February 01, 2023 Product: Amjevita The company has launched Amjevita (adalimumab), the first biosimilar referencing blockbuster drug Humira on the US market for certain serious inflammatory diseases.
Does 2022 already seem like a blur? The Pharma in Brief team is here for you with a summary of some of 2022’s most significant legal and regulatory developments in the Canadian pharmaceutical space. This left the PMPRB11 amendment, the validity of which was upheld by the FCA in December 2022 (see here ).
But that’s the controversy here: Did FDA approve LYTGOBI NDA 214801 on September 30, 2022 when the Agency issued its initial approval letter , or on October 5, 2022 when FDA issued a corrected approval letter ? Rather, “permission for commercial marketing” was only effective upon FDA’s issuance of its corrected October 5, 2022 letter.
On July 31, 2021, Health Canada published a Notice of Intent outlining its plan to amend the Food and Drug Regulations ( FDR ) and the Medical Devices Regulations in the spring of 2022. Generic drugs and biosimilars are not eligible for this application pathway. The amendments contemplate labelling flexibilities for special containers.
The manufacturer’s agreement must cover all its labeler codes that contain an applicable drug or a selected drug. All labeler codes that are covered by Discount Program agreements will be distributed to PDP sponsors and posted on the CMS website. had Part D expenditures on or before August 16, 2022). 1395w-114c(e).
Both India and Turkey are rising in this field, presenting significant trade and cooperation opportunities In 2022, the global pharmaceutical market reached a value of $ 1.5 The significant growth in biosimilars approvals and manufacturing further enhances patient access to biopharmaceuticals. trillion, with Turkey ranking 21st.
1 Used to treat a range of chronic diseases (eg, diabetes, rheumatoid arthritis, psoriasis, Crohn’s disease, haemophilia, etc), it is projected that the sales of biologics will rise from $380 billion in 2022 to $416 billion in 2023, and to almost $600 billion in 2027. 2022 [cited 2023Feb]. No serious adverse events were reported.
In 2022, Henlius signed a licensing and supply agreement with Organon, granting the latter with exclusive commercialisation rights for two biosimilar candidates, including HLX14. This agreement applies to markets such as the US, the European Union (EU), and Canada, with the exception of China.
While many states have had standing orders that allow for the dispensing of naloxone without an individual prescription, “harm reduction programs” that provide products and services to at-risk individuals still faced logistical difficulties in acquiring naloxone due to its prescription-only status which I discussed in this October 2022 blog post.
As we previously reported , Biogen sued Sandoz and Polpharma (“Defendants”) in a BPCIA litigation related to Defendants’ natalizumab biosimilar. On October 19, 2022, Biogen filed a Motion for Preliminary Injunction and Motion to Strike, while Sandoz filed a Cross Motion to Strike. Oral argument was held on May 17, 2023.
In 2022 the FTC issued special orders pursuant to Section 6(b) of the Federal Trade Commission Act (the “6(b) Orders” or “Orders”) to the six largest PBMs—Caremark Rx, LLC; Express Scripts, Inc.; OptumRx, Inc.; Humana Pharmacy Solutions, Inc.; Prime Therapeutics LLC; and MedImpact Healthcare Systems, Inc.
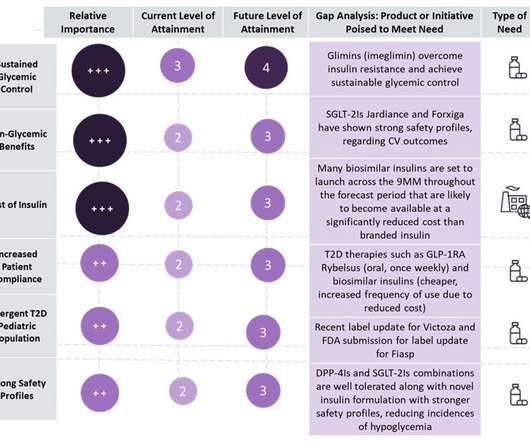
Type 2 diabetes (T2D) is a crowded and competitive landscape with multiple “me-too,” generic and biosimilar drugs entering the market, with market growth primarily driven by an increasing prevalent population across nine major markets (9MM: US, France, Germany, Italy, Spain, UK, Japan, China, and India).
The draft guidance follows the December 2022 enactment of the Food and Drug Omnibus Reform Act of 2022 (FDORA). FDA recommends manufacturers submit each modification, associated labeling changes and specific performance evaluation in the Modification Protocol (MP).
The long-awaited final rule, which we last discussed in a July 2023 blog post and have tracked in our March 2023 and March 2022 posts, aims to harmonize quality management system requirements for medical devices with requirements set forth by other regulatory authorities around the world. Notably, Part 820 will look different.
Users of eSTAR templates need to add attachments for cybersecurity risk management, cybersecurity management plan, continuing support plan, and cybersecurity labeling. implant programming), (4) software bill of materials, and (5) general labeling (connectivity and associated general cybersecurity risks, updateability/process).
The company faced a tough time in 2019 as the first biosimilars for its best-selling drugs (Rituxan, Avastin, and Herceptin) emerged. from 2018 to 2019 Roche’s blockbuster drugs, Herceptin, Avastin, and Rituxan played significantly well against its biosimilar products. Moreover, its medical device sales faced a decline of 1.7%
In the case of contract manufacturers, either the contract manufacturer or the person whose name appears on the label (i.e., The information is presented in a Q&A format. As reported previously , MOCRA includes a requirement for facility registration and listing of cosmetic products.
With the IRA, Congress changed the pricing model for certain “high-priced” Medicare-covered drugs without generic or biosimilar competition. Drugs are considered to be “qualifying single-source drugs” if they have been without generic or biosimilar competition for a certain number of years.
The last Warning Letter OPDP issued was in early 2022 to CytoDyn for promoting its investigational drug as an effective treatment for COVID-19 despite a failed study (and despite an FDA release issued a year prior about the same failed study – but that’s another story ). Clearly, OPDP thinks differently.
“All In” Manufacturer Definition CMS believes that the National Rebate Agreement (NRA) requires a “manufacturer” to report to Medicaid all of its covered outpatient drugs, under all of its labeler codes. This includes newly acquired labeler codes and newly formed subsidiaries.
We are unable to determine whether the number of such Warning Letters issued during the entire year represents a significant increase over 2022. The FDA compliance data, sorted by category, shows there were 159 Warning Letters issued to drug manufacturers or sponsors during 2023, with 161 issued in calendar year 2022. Terragene S.A.,
In fact, the priority designation for the final rule is labeled as “economically significant.” Jeff Shuren, director of FDA’s Center for Devices and Radiological Health (CDRH), during his remarks at the annual Food Drug and Law Institute (FDLI) conference in May 2023.
Most recently, Jeff Shuren, CDRH Director, gave a speech at the 2023 Food and Drug Law Institute (FDLI) annual conference in which he identified facilitating “availability of and access to existing and novel home-use medical technologies” as a strategic priority for 2022-2023 to advance health equity. lay users). seeking treatment).
As background, FDC Act § 505(q) states that FDA shall not delay approval of a pending ANDA, 505(b)(2) application, or 351(k) biosimilar application as a result of a citizen petition submitted to the Agency pursuant to 21 C.F.R. 1067, which first reared its head in Spring 2022 as an amendment to S.4348,
Serial testing was defined as testing symptomatic individuals twice over three days with at least 48 hours between tests, which is in accordance with study findings from the National Institute of Health referenced here and current EUA labeling for serial testing. Customer Support Help line).
However, as with drugs, dietary supplements require evidence that they are safe and that claims on product labels are truthful and not misleading. 2022 [cited 2024May]. 1 For use as prescription drugs, a botanical product must be approved by the FDA: to date, only two have gained this approval. researchspace.auckland.ac.nz.
Notably, this decision marks the first use of the new expedited procedures for withdrawal of an accelerated approval that were enacted in the Food and Drug Omnibus Reform Act of 2022 (FDORA). In December 2022, FDORA was enacted. Among other things, it revised the provisions relating to accelerated approval at 21 U.S.C. §
On October 18, 2022, CVM held a virtual listening session on the regulation of animal foods with certain types of claims. Furthermore, the label for the zootechnical animal food substance would need to include the statement: “Not for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animal.”
Performance Metric FY 2018 FY 2019 FY 2020 FY 2021 FY 2022 Average Number of FDA Days to MDUFA IV Decision 72.62 For labeling documents and the 510(k) summary, FDA often requests both clean and redlined versions. Average Number of Industry Days to MDUFA IV Decision 54.69 Average Number of Total Days to MDUFA IV Decision 127.31
It finalized a draft guidance published in 2022. Lewis, Senior Regulatory Device & Biologics Expert — On October 20, 2023, FDA announced the availability of the final guidance authored by CBER titled “Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies.”
at current exchange rates In Oct 2022, the acquisition was initiated and the transaction is expected to be adjusted per share by $0.07 Date - Jan 04, 2023 Product – N/A The acquisition is completed from a shareholder group led by Nordic Capital, in an all-cash transaction valued at $2.8B events/100 patient-yrs.) The authorization incl.
3 ,4, 5 As such, the NHC supports CMS proposal to ensure that Part D sponsors cover AOMs for obesity with clinical criteria that is not more restrictive than the FDA labeling for each AOM. Increased transparency will ensure that formulary practices align with broader goals of affordability and patient access.
Biden Administration HHS Budget Would Permit Third-Party Substitution of All Biosimilars On March 11th, the Biden Administration released its FY25 HHS Budget. and Europe alike, prescribers can already substitute any biosimilar for its reference product. Currently there are 10 biosimilars that can be substituted by U.S.
Terazosin - Off-label use in ureteral stone(s) expulsion Topical Corticosteroids - Apply thinly on the skin using fingertip unit. Statins - Not all statins need to be taken at night.
Even though President Joe Biden had asked the Secretary of HHS and the Attorney General to “initiate the administrative process to review expeditiously how marijuana is scheduled under federal law” in October 2022 , we also wondered how the Department of Justice (“DOJ”) and DEA would receive the HHS recommendation. Basis at 63-64.
Livornese — The Food and Drug Omnibus Reform Act (“FDORA”), enacted in December 2022, added a requirement that sponsors submit Diversity Action Plans (“DAPs”) for certain clinical studies involving drugs, biological products, or devices (codified at 21 U.S.C. § Would any such failure to meet DAP goals be reflected in labeling?
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content