This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
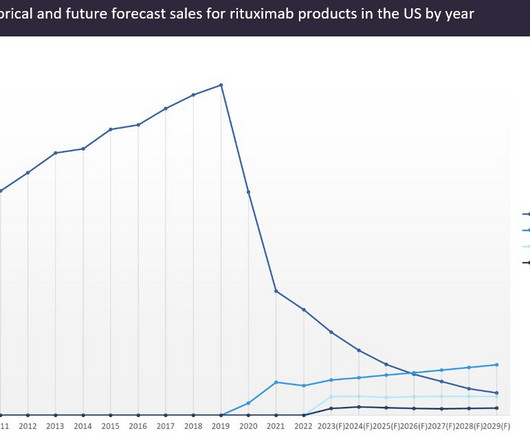
Rituxan received its first FDA approval in 1997 for the treatment of B-cell non-Hodgkin’s lymphoma (B-NHL) and reached peak global sales of $7.5bn in 2014. Rituxan’s success made it a prime target for biosimilar developers. Globally, the first rituximab biosimilar, Dr Reddy’s Laboratories’ Reditux, was approved in India in 2007.
The Food and Drug Administration (FDA) approved an average of 43 drugs in 2014-18, compared to an average of 23 in the first decade of the century. The biosimilar market in India is estimated to grow at 22 per cent CAGR to become $12 billion by 2025.
Because anabolic steroids are also abused to enhance athletic performance and increase muscle strength, Congress has enacted three laws regulating anabolic steroids: the Anabolic Steroid Control Acts of 1990 and 2004, and the Designer Anabolic Steroid Control Act of 2014 (“DASCA”). DASCA became law, amending the CSA, on December 18, 2014.
6 “The Biosimilar Red Tape Elimination Act”, which would prevent the HHS Secretary from requiring switching studies in order for a biosimilar to be deemed “interchangeable” Under U.S. state law, only biosimilars which are interchangeable may be substituted by a pharmacist without contacting the prescriber.
In the generic filers view, the regulatory review period should have started in January 2014 when the patent was reissued rather than in April 2004, when the IND was opened, because the reissued patent did not exist in April 2004. The plain text of 35 U.S.C.
A biosimilar is a biological medicine ‘similar’ to another biological medicine already approved in the EU (the ‘reference medicine’). 1 However, as most biosimilars are developed by means of a complex biotechnological process in living organisms, this inevitably leads to differences between the biosimilar and the reference product.
The Tropical Disease PRV program was born the very next year as part of the Food and Drug Administration Amendments Act of 2007 (FDAAA) and expanded in 2014 , which experts heralded as a game-changer for tropical disease therapies.
Cambridge, UK-based Coalesce, first formed in 2009 primarily as a contract services business, changed direction in 2014 to develop its own, in-house inhaler technologies. The post Sandoz bulks up ahead of possible sale with Coalesce buy appeared first on.
Xolair (omalizumab) has been a mainstay of treatment for years, and approval for the condition in 2014 was responsible for a leap in revenues for the drug – originally developed for a form of severe asthma – to blockbuster levels. Novartis reported $1.4 billion from the drug. .
He noted that the number of biotech startups has risen from just 50 in 2014 to over 5,000 now, reflecting India’s increasing focus on bioeconomy. He put forth the importance of Research and Development and regulatory systems, particularly for the deeper work needed in the biologics and biosimilar space.
Changes to regulatory data protection periods are of particular interest to biopharma” Changes to regulatory data protection (RDP) periods are of particular interest to biopharma (originators and generic / biosimilar manufacturers). By contrast, orphan drugs could become less profitable, more risky investments.
” Since 2014, the repayment percentage detailed in the VPAS and predecessor the PPRS average at around 7%, before skyrocketing to 19% in 2022 ahead of a government agreed cap at 15%. “The industry now regards the current approach, due to end in 2023, as broken.” The increase to 26.5% VPAS tax on top.
Previous iterations of this guidance from 2009 and 2014 (blogged on here and here ) were known as Good Reprint Practices (GRP). The SIUU Guidance, which supersedes the 2014 Draft GRP Guidance, is not substantially different in that regard and contains similar information as to the types of disclosures previously recommended.
In 2023, Rani Therapeutics plans to begin three additional Phase I studies with pipeline molecules RT-105 (containing an adalimumab biosimilar), RT-110 (containing PTH for hypo-parathyroidism) and RT-111 (containing an ustekinumab biosimilar for psoriatic arthritis, ulcerative colitis, Crohn’s disease and psoriasis).
When considering the expense of ERTs and SRTs, where biosimilars face difficult market penetration due to strict regulations, this will slow market growth. GlobalData’s Analyst covering Neurology and Ophthalmology, Thomas Parker, MPharmacol, predicted that Belsomra would lead the market by 2023.
The 2019 VPAS , just like its predecessor that was in place from 2014, has at its heart a payback mechanism. One of those is Celltrion Healthcare, which says that it is no longer viable to continue to supply the NHS with biosimilars. Predictable for government. The other company is not named.
We recount a few milestones along the road here: 1997 ASR Rule restricts sale, distribution of analyte specific reagents; FDA asserts authority over LDTs but articulates enforcement discretion policy 1998 FDA denies Citizen Petition submitted in 1992 on behalf of several clinical laboratories challenging agency authority over “home brew” tests 2006 (..)
Plaintiffs performed the necessary studies on BRAVECTO and filed an NADA on April 8, 2014; FDA approved the NADA on May 15, 2014. In February 2010, an oral formulation of BRAVECTO, a medication to treat and prevent fleas and tick infestation in dogs, was the subject of an INAD submitted to the Agency.
In 2015 the company also paid $17 billion to acquire Hospira , a firm specialising in injectable drugs and biosimilars, at a time when copycat biologics were starting to make real waves in the market. In 2014 the company made an offer of around $100 billion to acquire UK firm AstraZeneca (which at the time was going through a rough patch ).
Background FDA first introduced the final rule and guidance on eMDR in 2014 , which we blogged about here. Those who use eSubmitter will notice changes in the electronic 3500A template in the first week of March.
4,6 Five patent issues that European biosimilar developers should consider before entering the US market Quality control of the botanical drug substances was one of the critical factors that led to barricades during the development of Veregen and Mytesi. Available from: [link] Apply for a traditional herbal registration (THR) [Internet].
Eli Lilly’s Humalog compound and formulation patents expired in 2013 and 2014, respectively. In late 2017, the FDA approved Admelog, Sanofi’s biosimilar of Humalog. 6 The stakes were extremely high, with annual costs of insulin reaching $736 per patient in 2013, up threefold since 2002.
To better understand FDA’s approach in classifying postmarketing pregnancy studies as PMRs or PMCs, we reviewed all postmarketing requirements (PMRs) and postmarketing commitments (PMCs) related to maternal and fetal outcomes in FDA’s PMR/PMC database for drugs approved in the ten-year period from January 2014 through December 2023.
In 2014 Vanda introduced Hetlioz (tasimelteon) capsules to treat a rare sleep disorder. Examples in the regulation include extended-release formulations, new strengths, dosage forms, routes of administration, ingredients, and combinations. See our memo summarizing CMS’s regulation here.
In September 2014—nearly ten years ago!—PTE But there are still a couple of possibilities lurking out there! The first multiple PTE case up-to-bat concerns Gilead Sciences Inc.’s The first multiple PTE case up-to-bat concerns Gilead Sciences Inc.’s PTE applications were submitted to FDA for each of U.S. RE 44,599 and U.S.
Abbott Laboratories spun off its branded pharmaceutical business into Abbvie Amgen acquired Onyx Pharmaceuticals to boost its portfolio of Immuno-Oncology Johnson & Johnson attained strong growth in 2014 with momentum in their pharmaceutical business and strong sales of their new core products contributing $32.2B to the revenue.
In 2014, FDA approved Afrezza, which is a rapid-acting insulin that is taken before meals Glucagon Administration: In case of hypoglycemia/ low blood sugar, glucagon is administered to diabetes patients Moreover, based on the ability of insulin to infuse into the system, there are six groups or types as depicted in table 1 below.
. - Drug registration number Medicines (Advertisement & Sale) Act 1956 and Regulations - To learn prohibitions of medicine advertisement and Medicine Advertisement Board Registration of Pharmacists Act 1951 and Regulations - Pharmacy Board Malaysia and legislation aspects of provisional, full and temporary registration Code of Ethics for Pharmacists (..)
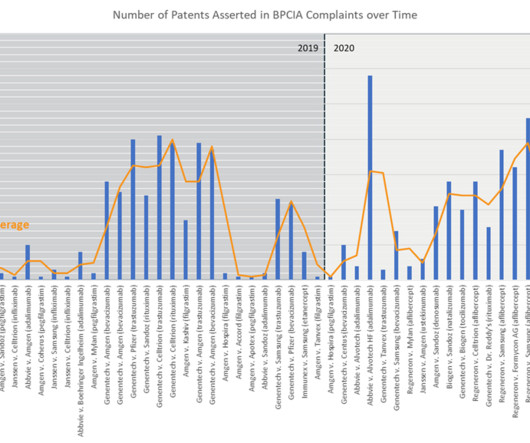
With 10 years of litigation since the first BPCIA complaint on NEUPOGEN (filgrastim) was filed in 2014, trends are becoming apparent that should cause all biopharma companies to reassess how they protect biologics and plan for biosimilar launch.
On October 24, 2014, Amgen initiated the first litigation under the Biologics Price Competition and Innovation Act (BPCIA), asserting infringement of two patents. They recognize that larger patent portfolios have proved valuable in delaying biosimilar competition. 262 (l)(5)(A) and (B)(ii). Amgen Inc. , 1, 9, 10, 16 (2017).
Since that time, it was formalized in FDA regulations (21 CFR 314 Subpart H) in 1992, codified in the Food, Drug, & Cosmetic Act by FDAMA (21 USC 356(c)) in 1997, revised by FDASIA in 2012, and described in guidance, most importantly, in the 2014 Expedited Programs for Serious Conditions Drugs and Biologics (2014 Guidance).
For instance, the December 2024 guidance goes beyond the 2014 Expedited Programs for Serious Conditions Drugs and Biologics guidance (the last to deal substantively with accelerated approval) to describe FDAs expectation that sponsors take a proactive approach to ensuring confirmatory trials are completed within specified timelines.
2014); U.S. The District Court in this case, which came to the opposite conclusion and is now reversed, was a notable outlier in a string of cases in other circuits upholding FDA’s authority to regulate stem cell clinics on similar grounds as the Ninth Circuit did here. Regenerative Sciences, LLC , 741 F.3d 3d 1314 (D.C. 3d 1302 (11th Cir.
59
59
Input your email to sign up, or if you already have an account, log in here!
Enter your email address to reset your password. A temporary password will be e‑mailed to you.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








































Let's personalize your content