This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Back in 2004, Gilead explained its decision by maintaining the medicines were not sufficiently different, but internal documents produced in court suggested the company made this move in order to maximize profits.
Back in 2004, Gilead explained its decision by maintaining the medicines were not sufficiently different, but internal documents produced in court suggested the company made this move in order to maximize profits. Continue to STAT+ to read the full story…
Children 29 days or older with fever from a documented viral source can be managed according to their clinical presentation and can go outside the algorithm. This requires a documented positive viral swab and not just a presentation consistent with a viral syndrome. 2004 Mar 10;291(10):1261-2. References: Roberts KB.
The assessment report of the Committee for Medicinal Products for Human Use (CHMP)’s Article 5(3) of Regulation (EC) No 726/2004 opinion on nitrosamine impurities in human medicinal products offers guidance and recommendations on mitigation and prevention of nitrosamine-contaminated human medicinal products.
2 The prerequisite to present statistically significant evidence for having efficacies of clinical relevance is crucial because FDA requires “adequate and well-controlled” multicentre clinical studies on any new drug candidate to document and support its safety and efficacy, and imposes the maximum level of scrutiny prior to approval.
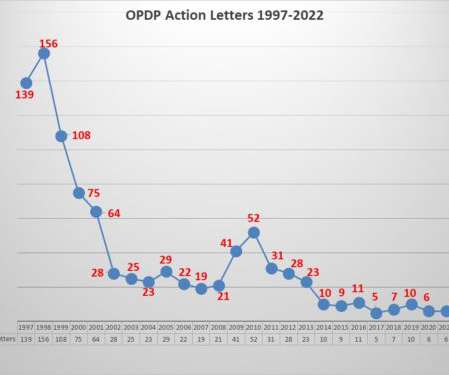
There are two means of gaining insight into the agency’s thinking about regulatory issues related to promotional communications by pharmaceutical companies; one is through the issuance of guidance documents, the other is through enforcement. But when it comes to enforcement things have changed greatly over the years.
This review will include key documents such as minutes of management review meetings and quality metrics which will enable them to quickly assess the company’s approach to quality and compliance and the current level of control. In addition, she worked as head of inspectorate and licensing for the MHRA from 2004-2006.
It is more helpful for students to individually outline solutions and then get together to work through all the details and compile the final document to submit. 2004; 2: 9–34. It is also helpful to let students know that dividing and conquering work is not beneficial as they need to know all information for future assessments.
He is an awe-inspiring peronality who has exceptional experiences in building and leading Institutional Sales, Key Account Management , Strategy & Planning , Market Access, Tender Business, New Product Inclusion, Document & Pricing strategy and much more.
The two groups were also separated by a sizeable time difference, with data from the control group ranging between 1990 and 2015, while Study 03-133 started enrolling patients in 2004. Changes in standard and supportive care over the time period could also have skewed the results, said the FDA reviewer.
Industry representatives spent many hours during 2004 discussing the needs of nursing home residents and the current standards of service. The document also requires plans to provide a transition period for non-formulary drugs to ensure beneficiaries have continuous access to required medications.
But the EU draft document says that the changes will make medicines more affordable, improve unequal access to medicines across the EU, and fulfil unmet patient needs.
In October 2023, the European Parliament (EP) proposed revisions to the European Commission proposals with diverging views on various topics set out through two documents. Figure 2: European Commission proposed changes to regulatory data protection periods for non-orphan and orphan drugs (Source: L.E.K) What are the latest amendments?
In 2009, Tysabri was performing well, having yielded $776 million in sales after being first approved in 2004, as per Biogen’s 2009 financial filings. These, along with other similar claims, meant Biogen owed potential damages of $1,036,900,151 to the US and the various States, as per court documents.
FDA briefing documents that address these reformulation strategies should include a description of the formulation design strategy that is employed to minimise the formation of NDSRIs in the drug product, including supporting manufacturing information and, at a minimum, three months of accelerated stability data demonstrating control of NDSRI.
CIPO’s application fees – and “small entities” – are getting larger in 2024 On January 1, 2024, CIPO imposed the first substantial increases to its service fees since 2004 (see here ). Draft regulations are expected to be released for consultation in 2024. The fee increases apply to both patent and trademark applications.
SoHO entities must implement a traceability system to unmistakably link each SoHO donor to their SoHO donation and to all documents, samples, SoHO preparations and SoHO entities that are associated with that SoHO from the point of collection to human application and outcome monitoring. Cited 2023Mar].
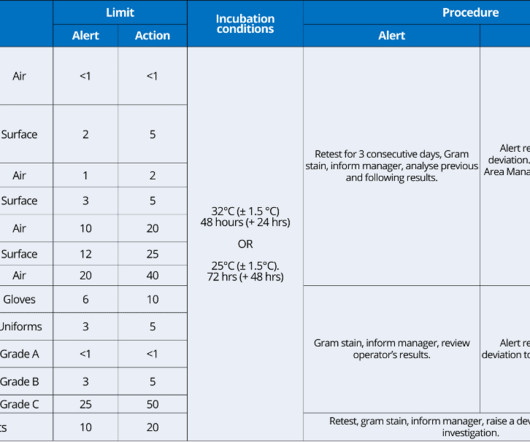
Environmental monitoring is a documented program which describes the routine particulate and microbiological monitoring of processing and manufacturing areas. Additional documents included each month. Additional documents included each month. Table of Contents What is meant by environmental monitoring? Checkout sample preview s.
A study in 2004 found an association between the presence of a mood or anxiety disorder, and the presence of anti-TPO antibodies. [1] Published 2004 Aug 18. She had already seen some major improvements with her Hashimoto’s through the use of thyroid medications as well as eating a Paleo diet , yet her anxiety persisted.
Increased transparency about data collection methods, including detailed metadata and documentation on how data is collected, processed, and any limitations, also plays a key role in facilitating access and use. Retrieved from https ://www.cms.gov/files/document/cmsmodernizinghealthcare.pdf 13 Zhang, S., 1, 2, and 3). Grenwelge, C.,
Fortunately, my colleague Dr. Kirk Gair from West Covina, CA, who is also a Hashimoto’s patient, has used cold lasers in his clinic since 2004 and has developed protocols that combine LLLT with chiropractic modalities. Remission of Hashimoto’s thyroiditis in a twelve-year-old girl with thyroid changes documented by ultrasonography.
A 2015 case report documented the thyroid labs of a man who had recurrent hyperthyroid episodes after three of his wife’s pregnancies! Published 2004 Aug 18. Like women, men can develop hormonal changes after the birth of a child, which can be linked to postpartum depression. [14] His wife had Hashimoto’s.) BMC Psychiatry.
Questions and Answers Regarding Food Allergens, Including the Food Allergen Labeling Requirements of the Federal Food, Drug, and Cosmetic Act This final guidance replaces previous draft and final guidance documents on food allergen labeling that FDA issued in November 2022, which we discussed in a previous post. By Sophia R.
In 2020, the Federal Trade Commission issued a report regarding settlements reached between brand and generic manufacturers in FY 2017 and noted that “for the first time since [FY] 2004, no settlement agreement in [FY] 2017 contains a no-[authorized generic] AG commitment.”. Throughout 2022, Novartis was at the opposing ends of legal cases.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





























Let's personalize your content